

 |
di: Diego Rosa |
Stabilità instabilità atmosferica
Parte settima
 Fig 1 - Amedeo Avogadro
Fig 1 - Amedeo AvogadroTorino, 9 agosto 1776 – Torino, 9 luglio 1856),
1) dQ = dU+dL
dQ e dL non sono differenziali di funzioni di stato mentre lo è dU, differenziale dell’energia interna U per cui si dovrebbe scrivere più correttamente:
2) δQ = dU+δL
indicando con δ non già l’incremento di una funzione di stato Q od L che non esistono, ma semplicemente delle forme differenziabili integrabili lungo una curva termodinamica. Per un gas δL = PdV e la 2) porge:
2’) δQ = dU+PdV
L’energia interna U, funzione di stato di due delle variabili P,V,T, per i gas ideali o “perfetti”, è funzione della sola temperatura T e si ha:
3) (dU/dT)V= nCV
Con CV calore specifico a volume costante del gas ed n numero di moli di gas .
Ricordiamo qui che una mole è un numero di particelle (atomi, molecole..) pari al numero di Avogadro, N0 ~ 6,02 1023 .
La fondamentale relazione di stato per i gas perfetti porge:
4) PV = nRT
Con n numero di moli di gas, R costante universale, pari 8,31 J/(molK )
La 4) porge: P =n/V RT: la temperatura assoluta è proporzionale alla pressione attraverso la densità delle particelle = n/V.
La relazione 2) indica che il calore ed il lavoro assorbiti (o ceduti) producono una variazione dell’energia interna, ma il loro contributo singolo non è a priori identificabile data una variazione di U. Q ed L non sono funzioni di stato; lo è la loro somma, U, come peraltro la temperatura T.
Considerazioni statistiche consentono di collegare la temperatura assoluta T di un gas perfetto all’energia cinetica media delle molecole Ec attraverso la relazione, data per No molecole:
5) Ec = fkTN0/2
Dove f è il grado di libertà della molecola (= 3 per le monoatomiche, 5 per le biatomiche), k la costante di Boltzmann = 1,38 10-23 J/K, k=R/N0 con R = costante dei gas = 8,31 J/molK , N0 il numero di Avogadro = 6,026 1023
2- Secondo principio
Il primo principio evidenzia l’energia interna U come variabile di stato funzione lineare della temperatura e stabilisce che in ogni trasformazione l’energia totale si deve conservare (impossibilità del moto perpetuo di prima specie ).
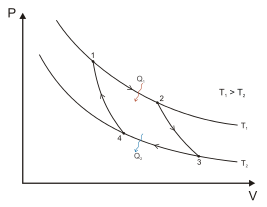 Fig. 2 - Il ciclo di Carnot
Fig. 2 - Il ciclo di Carnot
Q1/T1 = Q2/T2 e L = Q1-Q2 , η = (Q1-Q2)/Q1 = 1-T2/T1
Durante il ciclo reversibile di massimo rendimento (teorico) si ha tenendo conto dei segni che la somma Q1/T1+ Q2/ T2 = 0. La grandezza Q/T è allora una funzione di stato chiamata da Clausius S = Entropia . In termini differenziali dS = dq/T
Ogni ciclo può essere indefinitamente approssimato da una serie di cicli di Carnot e si ha:
6) ∮dQ/T = ∮dS = 0
Riprendiamo la 1) che per i gas diventa.
7) dQ = dU+pdV
e dividiamo per T otteniamo con dU = Cv dT:
8) dS = Cv dT+PdV/T
Esprimendo P in funzione di V mediante PV = RT abbiamo
9) dS = Cv dT+RT/TV dV = Cv dT+R/V dV
Le derivate miste di S, sono uguali:
10) d Cv/dV = d(R/V)/dT = 0
Per l’analisi ciò indica che dS è un differenziale esatto e S è una funzione di stato , funzione individuata univocamente dalle variabili di stato.
Per ogni trasformazione di un sistema in un ciclo chiuso, vale la relazione di Clausius:
11) |
|
essendo dQ positivo quando assorbito dal sistema |
L’uguaglianza valendo solo per i cicli ideali.
Ipotizziamo che un sistema termodinamico subisca delle trasformazioni interne rimanendo esso termicamente isolato verso l’esterno, trasformazioni che lo portano dallo stato A allo stato B. Riportiamo ora lo stesso sistema, non più isolato ed in modo reversibile dallo stato B allo stato A.
Per la relazione di Clausius abbiamo:
![]()
Ma il primo integrale è nullo perché il sistema è isolato mentre il secondo indica l’opposto dell’aumento di entropia. Si ha così:
![]()
 Fig 3 - Max Planck, Kiel 1858 - Gottingen
1947.
Fig 3 - Max Planck, Kiel 1858 - Gottingen
1947.Premio Nobel per la fisica 1918
L’entropia del sistema è aumentata.
L’intero universo considerandolo chiuso ed isolato aumenterà indefinitamente.
Considerazioni di meccanica statistica hanno portato L. Boltzman
a considerare l’entropia di un sistema chiuso ed isolato come funzione del
numero
Ω dei
microstati (molecolari) corrispondenti ad un singolo macrostato funzione
definita successivamente da M. Planck come
S= k logΩ, dove
k è una
costante universale pari a 1,38 10-23. Se il sistema non è isolato
S è funzione
della distribuzione di probabilità dei singoli microstati.
3. Il terzo principio
A queste due leggi o principi si associa anche il terzo principio.
Nella maggior parte delle applicazioni della termodinamica conta la variazione di entropia non già il suo valore assoluto. Tuttavia le considerazioni di meccanica statistica indicano di considerare ad entropia S = 0 il solido monocristallino alla temperatura (teorica) dello zero assoluto. In questo caso i microstati si ridurrebbero ad uno essendo le molecole indistinguibili anche dal punto di vista energetico.